major areas of research
Evolutionary couplings:
Predicting protein structures, complexes, and conformational plasticity from evolution
Over the past 2 years we have been applying a comparative genomics approach to the classic problem of ab initio 3D structure prediction of proteins, based on the principle that amino acids undergoing correlated substitutions are likely close in structure. Although approaches based on this principle had been explored for over 20 years, a key insight was to understand that observed amino acid correlations in sequence alignments contain confounding transitivity - a well-known phenomenon in highly coupled networks. We use a global statistical model to disentangle causal from transitive correlations and then apply the resulting evolutionary couplings to predict protein structure. We have developed this evolutionary approach to accurately predict large pharmaceutically relevant membrane proteins (600 amino acids), the 3D structure of multi-protein complexes and, built a publicly available server, EVCouplings.org. Our lab is building on this work to determine the structure of large macromolecular complexes and to develop methods for detecting conformational plasticity.
Publications: Marks et al (2011), PLOS One 2011. Hopf et al (2012), Cell. Marks, Hopf, and Sander (2012), Nature Biotechnology. Hopf, Scharfe et al (2014), eLife.

rna:
Exploring evolutionary constraints on RNA
We will continue to critically appraise the cellular functions and evolutionary constraints of RNAs with the goal of aiding RNA-based molecular biological tools, therapeutics and our understanding of the co-evolution of protein and RNA. We published the first prediction method for determining microRNA targets that led to an early appreciation of the large number of genes that could be regulated by microRNAs, the combinatorial nature of microRNA regulation and the discovery of the first viral microRNA. Taking a more global approach we then discovered crucial determinants of si/microRNA targeting that are important for pharmacological development of small RNA therapeutics and the use of siRNAs as molecular tools. The unsolved mystery of the evolutionary constraints on microRNAs, together with the steady stream of yet more RNA species , e.g. lncRNAs, RNA in the CRISPR system, suggests genomic analyses of RNAs still has a long way to go.
Publications: Bino et al (2004), PLoS Biology. Khan et al (2009), Nature Biotechnology. Arvey et al (2010), Molecular Systems Biology. Schmiedel et al (2015), Science. Weinreb et al (2016), Cell.

order in disorder:
expanding our definition of protein folding
Protein flexibility ranges from simple hinge movements to functional disorder. Around half of all human proteins contain apparently disordered regions with little 3D or functional information, and many of these proteins are associated with disease. Building on the evolutionary couplings approach previously successful in predicting 3D states of ordered proteins and RNA, we developed a method to predict the potential for ordered states for all apparently disordered proteins with sufficiently rich evolutionary information. The approach is highly accurate (79%) for residue interactions as tested in more than 60 known disordered regions captured in a bound or specific condition. Assessing the potential for structure of more than 1,000 apparently disordered regions of human proteins reveals a continuum of structural order with at least 50% with clear propensity for three- or two-dimensional states. Co-evolutionary constraints reveal hitherto unseen structures of functional importance in apparently disordered proteins
Publications: Toth-Petroczy et al (2016), Cell.

functional consequences of mutation and protein design:
predicting how mutations will interact
Quantitative prediction of the effects of polymorphism and mutation remains a major challenge in biomedical research and protein engineering. As a starting point, our lab is developing methodologies that build on the observation that functional sites in proteins are often constrained by a mixture of independent conservation and dependent co-conservation. Our goals are to predict the effects of context dependent mutations, to account for epistasis between positions, and to predict the effects of more than one mutational change. By accurately predicting how mutations will interact, we are exploring the possibility of generating diverse libraries of novel sequences that retain functionality and stability and can be used in the directed evolution of biocatalysts and therapeutics.
Publications: Hopf et al (2017), Nature Biotechnology. Hopf et al (2015), hosted on arXiv.

genetic variation and drugs:
smart integration for understanding drug-gene interactions
Two biological realities confound the search for genetic causes of disease and pre-disposition to drug response. The first is that many different mutational events across a population can cause similar phenotypes. The second is that combinations of polymorphic events, in a single person, can contribute to disease likelihood and therapeutic response. Our lab is starting to address this by applying a “Benedict Cumberbatch" approach to finding more biologically informed priors and algorithms for reducing the combinatorial search space in associating combinations of factors with disease. To aid this effort we built an infrastructure that connects drugs, disease, genes and genetic variation in an information retrieval system called “dopey”.

theoretical developments:
improving inferences of undirected graphical models
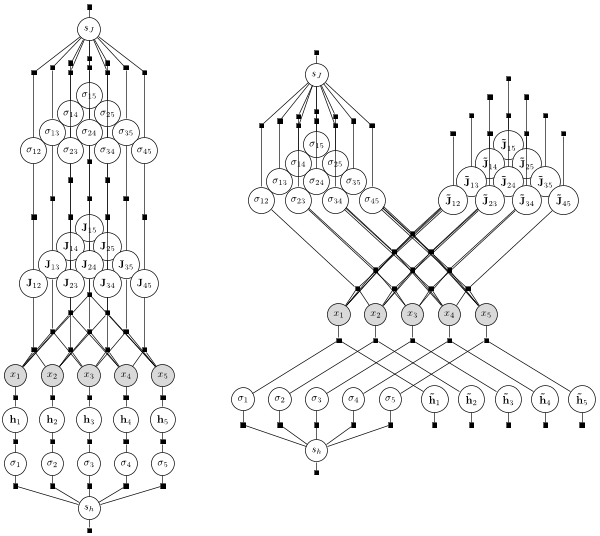
Bayesian approaches for single-variable and group-structured sparsity outperform L1 regularization, but are challenging to apply to large, potentially intractable models. Here we show how noncentered parameterizations, a common trick for improving the efficiency of exact inference in hierarchical models, can similarly improve the accuracy of variational approximations. We develop this with two contributions: First, we introduce Fadeout, an approach for variational inference that uses noncentered parameterizations to capture a posteriori correlations between parameters and hyperparameters. Second, we extend stochastic variational inference to undirected models, enabling efficient hierarchical Bayes without approximations of intractable normalizing constants. We find that this framework substantially improves inferences of undirected graphical models under both sparse and group-sparse priors.
Publications: Ingraham and Marks (2016), hosted on arXiv.